Open and FAIR science and scientific workflows
Overview
Teaching: 0 min
Exercises: 0 minQuestions
Key question (FIXME)
Objectives
First learning objective. (FIXME)
FIXME
Key Points
First key point. Brief Answer to questions. (FIXME)
Introduction to scientific workflow management systems
Overview
Teaching: 0 min
Exercises: 0 minQuestions
Key question (FIXME)
Objectives
First learning objective. (FIXME)
Workflow description
We will assemble a simple workflow easy to understand for anyone who attended basic biology classes. The goal of this workflow is to predict Open Reading Frames (ORFs) from RNA sequences and filter these predicted ORFs to keep only those containing the specific amino acids (sub)sequence “VERA”.
Although very simple, our implemetation will demonstrate:
- how to load remote data into Galaxy
- how to assemble multiple steps into a workflow
- how to use the split-apply-combine strategy
Here is a step-by-step overview:
- Step 1: download a fasta file containing RNA sequence from ensembl
- Step 2: split fasta file in eg 10 files (for parallel processing)
- Step 3: translate the sequence with an ORF finder (here I used a getorf wrapper on emboss)
- Step 4: filter for desired pattern
- Step 5: merge back translated fastq files
- Step 6: extract the original transcript IDs
Key Points
First key point. Brief Answer to questions. (FIXME)
Running the example in Galaxy
Overview
Teaching: 0 min
Exercises: 0 minQuestions
Key question (FIXME)
Objectives
First learning objective. (FIXME)
The Galaxy Platform
Galaxy is an outlier in the set of workflow management systems (WMSs) that we are going to discuss, because it includes a (web) user interface to design and run the workflows. It is Open Source under Academic Free License and developed at Penn State, Johns Hopkins, OHSU and Cleveland Clinic with substantial contributions from outside these institutions. The Galaxy Project includes several public serves, including:
- usegalaxy.eu
- usegalaxy.org
- usegalaxy.org.au
And several others. For this exercise, we are going to use usegalaxy.eu. First, let’s create a user there.
- Create an account in Galaxy
According to the developer, the core values of Galaxy are:
- accessibility (users without programming experience)
- reproducibility
- transparency
Overview of the Galaxy UI
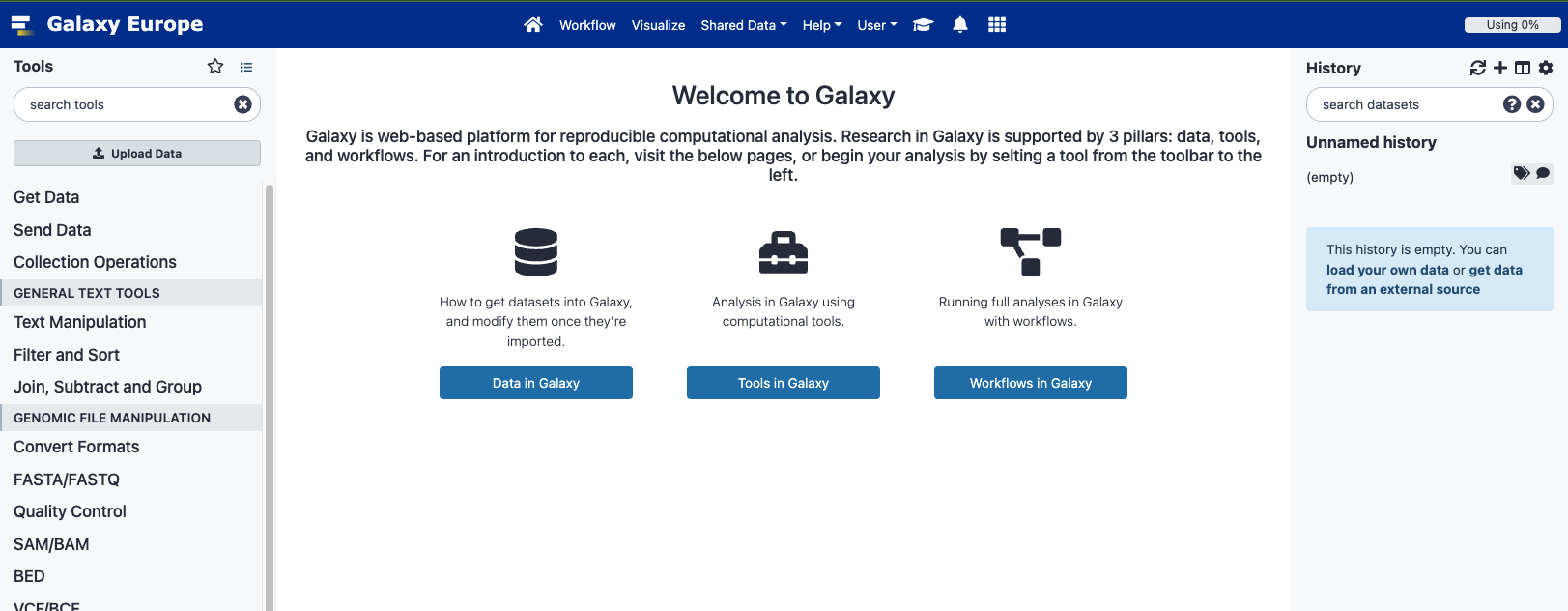
- On the left, available tools.
- In the center, main panel to run tools and view results.
- On the right, dataset history (where the results will accumulate).
Basic usage: first thing to do is to import a dataset, then search for the right tool (search bar), then execute it and the results will appear in the central panel. Job results accumulate in your History on the right.
Tools in Galaxy
The tools you run through Galaxy are the same tools that you can run through the command line.
Datasets in Galaxy
Dataset (imported files or result files) in history. If you click on a dataset name, you can download it, get a link to it, understand how much resource it is using etc. You can also rerun the dataset (learn the exact parameters) and visualise the dataset.
Shared Data menu allows you to access dataset for trials and training.
What happens behind the scenes
When run a workflow, Galaxy takes care of the job submission to HPC. Many jobs can run in parallel. Galaxy helps you visualizing the job dependencies and status:
- green: job successfully completed
- yellow: running
- grey: waiting, e.g. for a preliminary task to be completed
- red: failed
It’s Galaxy who takes care of figuring out the dependency and running all steps in the correct order.
Assembling and running the example Workflow in Galaxy
For this demo, we’ll use the public european galaxy server. If you would like to run this demo during or after the workshop, please create a free account. For this, please use the register link in the top menu, also directly available here.
Step-by-Step
Login in usegalaxy.eu
- Create a new History (if needed) and name it “ORF Detection”
Grab a cDNA FASTA link from ENSEMBL FTP web site
- Open a new tab in your browser
- Go to the ensembl web site, select human and click on the
Download FASTAlink available in theGene annotationpanel. This brings you to the FTP site - Navigate to the
cdnafolder and right-click on theHomo_sapiens.GRCh38.cdna.all.fa.gzfile to copy its URL (copy link address) - Go back to your galaxy tab and click the
Upload Dataat the top of theToolspanel (on the left). - In the pop-up, click the
Paste/Fetch dataon the bottom and :- Paste the copied FTP URL to the main text area:
http://ftp.ensembl.org/pub/release-105/fasta/homo_sapiens/cdna/Homo_sapiens.GRCh38.cdna.all.fa.gz - Give a name to the file:
Homo_sapiens.GRCh38.cdna.all.fa.gz - Set the type to :
fasta.gz - Set the Genome to: start typing
grch38and selectHuman Dec. 2013 ...(hg38) - Click
Startand thenClose
- Paste the copied FTP URL to the main text area:
=> a new dataset is created in your history. The grey color indicates that the job creating this dataset is not yet started. The dataset color will turn “salmon (?)” when the job will be executing to finally become green (or red) upon successful completion (or error)
Split the downloaded FASTA
For this we will use the faSplit tool
- Search for
faSplit(mind the case) in the Tools’ search box - Click on the
faSplit Split a FASTA filetool - Fill in the form:
- Input: the fastq file we just fetched
- Split by:
Number of files (for multi sequence FASTA) - Number of chunks:10
- Click
Execute
=> The result dataset is now a collection of FASTA files (for a weird reason 9 files were created)
Predict ORFs with the getorf tool
- Search for
getorforORFin the Tools’ search box - Fill in the form:
- Input: click on the
foldericon to select your fasta file collection - Use defaults for all other fields
- Input: click on the
- Click
Execute
=> The files are processed in parallel i.e. they are executed on different cluster node at the same time (if cluster load permits)
Filter FASTA on the requested motif
- Search for
Filter FASTA on the headers andin the Tools’ search box- the tool should be in the first 10 hits (yes the galaxy search is not the best)
- Fill in the form:
- Input: click on the
foldericon to select your getorf collection - Criteria for filtering on the sequences: select
Regular expression...- Add
VERAin the new Regular expression pattern the sequence should match test box
- Add
- Input: click on the
- Click
Execute
Merge back translated fastq files
- Search for
Concatenate datasets tail-to-headin the Tools’ search box- the tool should be in the first 10 hits (yes the galaxy search is not the best)
- Fill in the form:
- Input: click on the
foldericon to select yourFilter FASTAcollection
- Input: click on the
- Click
Execute
Optional Extract unique list of transcript (ENST)
Step 1: convert the fasta file to a tabular format
- Search for ` FASTA-to-Tabular converter` in the Tools’ search box
- Fill in the form:
- Input: select the merged dataset from previous step
- How many columns to divide title string into?:
2
- Click
Execute
=> the result is a table with 3 columns:
- The ENST ID in col 1
- The rest of the description in col 2
- The sequence in col 3
Step 2: reformat the accession number
- Search for
Column Regex Find And Replacein the Tools’ search box - Fill in the form:
- Input: select the table generated at previous step
- Click on insert Check
- Find Regex: paste
(ENST[0-9]+)\..+ - Replacement:
\1
- Find Regex: paste
- Click
Execute
Step 3: extract the first column
- Search for
Cut columns from a tablein the Tools’ search box - Fill in the form:
- Input: select the table generated at previous step
- Cut columns: paste
c1
- Click
Execute
Step 4: extract the unique list of ENST
- Search for
Unique occurrences of each record (Galaxy Version 1.1.0)in the Tools’ search box - Fill in the form:
- Input: select the table generated at previous step
- Click
Execute
Workflow creation/extraction
Use the Extract Workflow in the history options and adapt the name (to eg ‘VERA ORF Predition WF’) and click Create Workflow and immediatly click the edit link on the notification.
When extracting the workflow, only select the steps described above ie make sure to unselect any other computation you may have done.
Once in the workflow editor, review each step and make sure the option are identical to those above ; and all connections are preserved.
Then click run and select the fasta cDNA FASTA file and click Run
Key Points
First key point. Brief Answer to questions. (FIXME)
Running the example in Snakemake
Overview
Teaching: 0 min
Exercises: 0 minQuestions
Key question (FIXME)
Objectives
First learning objective. (FIXME)
Snakemake
GNU make + Python = Snakemake
GNU make: software to manage projects with several intermediate steps. “You only build what needs to be built” Instructions are written in a makefile, which looks like this:
all proteins.faa
proteins.faa: transcripts.fna
getorf -sequence transcripts.fna -outseq proteins.faa
Why Snakemake instead of make?
- Python-dialect
- Native support for reproducibility principles
- different environment
- conda/mamba singularity
- docker support
- HPC support
- Simple control-flow mechanisms
- Python API (bind Snakemake workflows into a Python application)
Assembling and running the example Workflow in Snakemake
Step-by-Step
Download transcripts
TRANSCRIPTS_URL = "http://ftp.ensembl.org/pub/release-105/fasta/naja_naja/cdna/Naja_naja.Nana_v5.cdna.all.fa.gz"
rule all:
input:
"output/transcripts.fa.gz",
rule fetch_transcriptome:
output:
"output/transcripts.fa.gz"
params:
url = TRANSCRIPTS_URL
shell:
"""
mkdir -p output/
wget {TRANSCRIPTS_URL} -O output/transcripts.fa.gz
"""
Filter by length
ENVIRONMENTS = "../envs"
Point at environments.
rule all:
input:
"output/transcripts.fa.gz",
"output/transcripts.length_filtered.fa.gz",
Add filtered to rule all.
rule filter_by_length:
input:
transcripts = rules.fetch_transcriptome.output.transcripts
output:
filtered = rules.fetch_transcriptome.output.transcripts.replace(".fa.gz", ".length_filtered.fa.gz")
params:
minlen = 300
conda:
f"{ENVIRONMENTS}/bbmap.yml"
shell:
"""
bbduk.sh in={input.transcripts} out={output.filtered} minlen={params.minlen}
"""
Splitting transcriptome (“scatter”)
Add the rule to split the transcriptome.
rule split_transcriptome:
input:
rules.filter_by_length.output.filtered
output:
txome_chunks = expand(os.path.join("output", "txome_chunks", "chunk-{chunk}.fa"), chunk=range(10))
params:
chunksize = 2935
shell:
"""
mkdir -p output/txome_chunks/
gzip -dc {input[0]} | awk 'BEGIN {{n=0;m=0;}} /^>/ {{ if (n%{params.chunksize}==0) {{f=sprintf(\"output/txome_chunks/chunk-%d.fa\",m); m++;}}; n++; }} {{ print >> f }}'
"""
Check the steps
This is currently the structure of your Snakemake file.
TRANSCRIPTS_URL = ... ENVIRONMENTS = ... rule all: input: ... rule fetch_transcriptome: ... rule filter_by_length: ... rule split_transcriptome: ...
Find and translate ORFs
rule find_and_translate_orfs:
input:
chunk = os.path.join("output", "txome_chunks", "chunk-{chunk}.fa"),
output:
orfs = os.path.join("output", "orf_chunks", "chunk-{chunk}.orfs.fa"),
params:
minlen = 300
conda:
f"{ENVIRONMENTS}/emboss.yml"
shell:
"""
mkdir -p output/orf_chunks/
getorf -sequence {input.chunk} -outseq {output.orfs} -minsize {params.minlen}
"""
Extract toxins
rule extract_toxins:
input:
toxin_proteins_blast = rules.combine_chunks_and_filter.output.toxin_proteins_blast,
proteins = rules.combine_proteins.output.all_proteins
output:
toxin_proteins = os.path.join("output", "toxins.faa")
conda:
f"{ENVIRONMENTS}/seqtk.yml"
shell:
"""
seqtk subseq {input.proteins} <(cut -f 1 {input.toxin_proteins_blast} | sort -u) > {output.toxin_proteins}
"""
Final result
# TRANSCRIPTS_URL = "http://ftp.ensembl.org/pub/release-105/fasta/naja_naja/cdna/Naja_naja.Nana_v5.cdna.all.fa.gz"
# ENVIRONMENTS = "../envs"
# report: "workflow_report.rst"
rule all:
input:
transcripts = "output/transcripts.fa.gz",
txome_chunks = expand(os.path.join("output", "txome_chunks", "chunk-{chunk}.fa"), chunk=range(10)),
orf_chunks = expand(os.path.join("output", "orf_chunks", "chunk-{chunk}.orfs.fa"), chunk=range(10)),
toxin_chunks = expand(os.path.join("output", "toxin_chunks", "chunk-{chunk}.toxins.tsv"), chunk=range(10)),
all_proteins = os.path.join("output", "all_proteins.faa"),
blast_output = os.path.join("output", "toxin_blast.filtered.tsv"),
toxins = os.path.join("output", "toxins.faa")
shell:
"""
rm -rvf {input.transcripts} $(dirname {input.txome_chunks}) $(dirname {input.orf_chunks}) $(dirname {input.toxin_chunks}) {input.all_proteins} {input.blast_output}
"""
rule fetch_transcriptome:
output:
transcripts = "output/transcripts.fa.gz"
params:
url = config["TRANSCRIPTS_URL"]
shell:
"""
mkdir -p output/
wget {params.url} -O {output.transcripts}
"""
rule filter_by_length:
input:
transcripts = rules.fetch_transcriptome.output.transcripts
output:
filtered = rules.fetch_transcriptome.output.transcripts.replace(".fa.gz", ".length_filtered.fa.gz")
params:
minlen = config["transcript_criteria"]["minimum_transcript_length"]
conda:
f"{config['ENVIRONMENTS']}/bbmap.yml"
shell:
"""
bbduk.sh in={input.transcripts} out={output.filtered} minlen={params.minlen}
"""
rule split_transcriptome:
input:
rules.fetch_transcriptome.output.transcripts
output:
txome_chunks = expand(os.path.join("output", "txome_chunks", "chunk-{chunk}.fa"), chunk=range(10))
params:
chunksize = config["transcript_criteria"]["chunksize"]
shell:
"""
mkdir -p output/txome_chunks/
gzip -dc {input[0]} | awk 'BEGIN {{n=0;m=0;}} /^>/ {{ if (n%{params.chunksize}==0) {{f=sprintf(\"output/txome_chunks/chunk-%d.fa\",m); m++;}}; n++; }} {{ print >> f }}'
"""
rule find_and_translate_orfs:
input:
chunk = os.path.join("output", "txome_chunks", "chunk-{chunk}.fa"),
output:
orfs = os.path.join("output", "orf_chunks", "chunk-{chunk}.orfs.fa"),
params:
minlen = 1000
conda:
f"{config['ENVIRONMENTS']}/emboss.yml"
shell:
"""
mkdir -p output/orf_chunks/
getorf -sequence {input.chunk} -outseq {output.orfs} -minsize {params.minlen}
"""
rule find_toxins:
input:
proteins = os.path.join("output", "orf_chunks", "chunk-{chunk}.orfs.fa"),
db = config["VENOM_DATABASE"]
output:
toxin_hits = os.path.join("output", "toxin_chunks", "chunk-{chunk}.toxins.tsv")
conda:
f"{config['ENVIRONMENTS']}/blast.yml"
threads:
2
shell:
"""
mkdir -p output/toxin_chunks/
blastp -db {input.db} -query {input.proteins} -num_threads {threads} -out {output.toxin_hits} -outfmt '6 qseqid sseqid pident qstart qend sstart send qlen slen length nident mismatch positive gapopen gaps evalue bitscore'
"""
rule combine_chunks_and_filter:
input:
expand(os.path.join("output", "toxin_chunks", "chunk-{chunk}.toxins.tsv"), chunk=range(10))
output:
toxin_proteins_blast = os.path.join("output", "toxin_blast.filtered.tsv")
params:
eval_threshold = config['transcript_criteria']['eval_threshold']
shell:
"""
awk '$16 < {params.eval_threshold}' {input} > {output.toxin_proteins_blast}
"""
rule combine_proteins:
input:
expand(os.path.join("output", "orf_chunks", "chunk-{chunk}.orfs.fa"), chunk=range(10))
output:
all_proteins = os.path.join("output", "all_proteins.faa")
shell:
"""
cat {input} > {output.all_proteins}
"""
rule extract_toxins:
input:
toxin_proteins_blast = rules.combine_chunks_and_filter.output.toxin_proteins_blast,
proteins = rules.combine_proteins.output.all_proteins
output:
toxin_proteins = os.path.join("output", "toxins.faa")
conda:
f"{config['ENVIRONMENTS']}/seqtk.yml"
shell:
"""
seqtk subseq {input.proteins} <(cut -f 1 {input.toxin_proteins_blast} | sort -u) > {output.toxin_proteins}
"""
Key Points
First key point. Brief Answer to questions. (FIXME)
Running the example in Nextflow
Overview
Teaching: 0 min
Exercises: 0 minQuestions
Key question (FIXME)
Objectives
First learning objective. (FIXME)
Nextflow
Core features:
- Fast prototyping
- Reproducibility
- Portability
- Simple parallelism
- Continuous checkpoints
Differences to snakemake:
- Don’t need to specify the names of output files/folder
- Groovy instead of Python
- Cannot do a dry run (would have to use small datasets)
Functions in workflows management system
Our example doesn’t cover this, so you will not see Python/Groovy flavour, but consider at a certain level of advancement this will be needed.
- Resource usage plots
- Nextflow tower
- nf-core (predefined pipelines)
Assembling and running the example Workflow in Nextflow
Step-by-Step
Final result
#!/usr/bin/env nextflow
nextflow.enable.dsl = 2
params.url = "http://ftp.ensembl.org/pub/release-105/fasta/ciona_intestinalis/cdna/Ciona_intestinalis.KH.cdna.all.fa.gz"
params.sampleid = "Ciona_intestinalis"
workflow {
input_ch = Channel.from(params.url)
fetch_data(input_ch)
length_filter(fetch_data.out)
gene_calling(length_filter.out.splitFasta(by:2000, file:true))
join_fastas(gene_calling.out.collect())
DEAD_motif_search(join_fastas.out, params.sampleid)
}
process fetch_data {
input:
val url
output:
path "*.fa.gz"
script:
"""
wget ${url}
"""
}
process length_filter {
container "https://depot.galaxyproject.org/singularity/seqkit%3A2.1.0--h9ee0642_0"
input:
path fasta
output:
path "length_filtered.fa"
script:
"""
seqkit seq --min-len 500 ${fasta} > length_filtered.fa
"""
}
process gene_calling {
container "https://depot.galaxyproject.org/singularity/prodigal%3A2.6.3--h779adbc_3"
input:
path fasta
output:
path "proteins.faa"
script:
"""
prodigal -i ${fasta} -a proteins.faa -o proteins.gbk
"""
}
process join_fastas {
input:
path "fasta?.faa"
output:
path "joined.faa"
script:
"""
cat *.faa > joined.faa
"""
}
process DEAD_motif_search {
container "https://depot.galaxyproject.org/singularity/seqkit%3A2.1.0--h9ee0642_0"
publishDir "results/${sampleid}/", mode: 'copy'
input:
path fasta
val sampleid
output:
path "dead_motif_containing_genes.faa"
path "ids.txt"
script:
"""
seqkit grep -s -p DEAD ${fasta} > dead_motif_containing_genes.faa
grep ">" dead_motif_containing_genes.faa | awk ' { print substr(\$1, 2)}' > ids.txt
"""
}
Key Points
First key point. Brief Answer to questions. (FIXME)